CHEM21 Case Study: One Pot Synthesis of N-substituted β-amino Alcohols
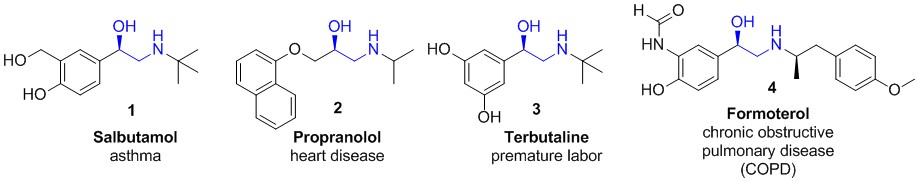
The β-amino alcohol moiety is a privileged structural motif in the pharmaceutical industry. Salbutamol (1) and propranolol (2) are on the World Health Organization List of Essential Medicines [1] and represent the most important examples of therapeutic agents bearing this structural feature. Numerous others have been released on the market for the treatment of various circulatory, respiratory and other diseases (Figure 1). In addition to their high relevance in drug discovery,[2][3][4] N-substituted β-amino alcohols are important building blocks in the preparation of added value chemicals [5][6] and ligands for catalysis.[7][8]
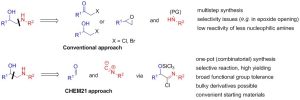
The construction of the β-amino alcohol fragment is almost invariably achieved by the nucleophilic attack of an amine on a suitable electrophilic reaction partner, such as an epoxide, α-haloketone or β-halohydrin (Scheme 1). Although robust, there are many drawbacks associated with this synthetic strategy. First, the required starting materials are typically not commercial and their multistep preparation is highly wasteful and time-consuming. Secondly, the substitution approach poses important selectivity issues (e.g. regioselectivity in the epoxide opening), double alkylation of the (unprotected) primary amine and low reactivity of poorly nucleophilic and bulky amines which thus need to be employed in large excess. A general alternative strategy circumventing these problems and employing readily available building blocks would therefore be a valuable tool for medicinal as well as process chemistry.
CHEM21 researchers designed a novel method for the construction of the N-substituted β-amino alcohol motif via a less straightforward central C-C bond retrosynthetic disconnection through addition rather than substitution, using aldehydes and isocyanides as building blocks. Aldehydes, isocyanides and SiCl4 can be combined in a Passerini-type reaction to give α-trichlorosilyloxy imidoyl chlorides,[10][11] which are versatile intermediates towards the synthesis of valuable compounds like α-hydroxy amides and α‑hydroxy esters. For the synthesis of β-amino alcohols, the CHEM21 researchers employed in the same pot the generation of the imidoyl chloride intermediate from aldehydes and isocyanides in combination with its reduction with a mild reducing agent (ammonia-borane). This approach furnishes the desired β‑amino alcohol derivatives in a faster, simpler and more general way than current methodology (Scheme 2).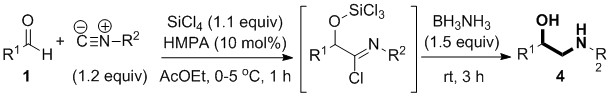
The synthetic utility of this method was validated by the preparation of approx. 40 β-amino alcohols in good yields (44-93%). Importantly, high performance is achieved in challenging circumstances for conventional approaches: oxidation-sensitive functional groups, bulky derivatives, nucleophilic substitution-sensitive substrates and non-nucleophilic amines. Furthermore, the method can be upgraded to a catalytic enantioselective synthesis by introducing a (commercial) chiral catalyst.
The good availability of the required building blocks, the reduced reaction time and the general scope recommend this method for both combinatorial and medicinal chemistry applications.[9]
- WHO essential medicines, 22nd edition (Last accessed: August 2022).
- , Flavone-Based Novel Antidiabetic and Antidyslipidemic Agents, Journal of Medicinal Chemistry, 2012, 55, 4551-4567.
- , Novel Carvedilol Analogues That Suppress Store-Overload-Induced Ca2+ Release, Journal of Medicinal Chemistry, 2013, 56, 8626-8655.
- , Hybrids consisting of the pharmacophores of salmeterol and roflumilast or phthalazinone: Dual β2-adrenoceptor agonists-PDE4 inhibitors for the treatment of COPD, Bioorganic & Medicinal Chemistry Letters, 2013, 23, 1548-1552.
- , Discovery of novel bis-oxazolidinone compounds as potential potent and selective antitubercular agents, Bioorganic & Medicinal Chemistry Letters, 2014, 24, 1496-1501.
- , cis-1-Oxo-heterocyclyl-4-amido cyclohexane derivatives as NPY5 receptor antagonists, Bioorganic & Medicinal Chemistry Letters, 2014, 24, 1458-1461.
- , Achiral β-amino alcohols as efficient ligands for the ruthenium-catalysed asymmetric transfer hydrogenation of sulfinylimines, Tetrahedron Letters, 2011, 52, 789-791.
- , Enantioselective addition of diethylzinc to aldehydes catalyzed by β-amino alcohols derived from (1R,2S)-norephedrine, Applied Organometallic Chemistry, 2010, 24, 33-37.
- , One-Pot Synthesis of N-Substituted β-Amino Alcohols from Aldehydes and Isocyanides, Chemistry – A European Journal, 2015, 21, 7808-7813.
- , The First Catalytic, Asymmetric α-Additions of Isocyanides. Lewis-Base-Catalyzed, Enantioselective Passerini-Type Reactions, Journal of the American Chemical Society, 2003, 125, 7825-7827.
- , Catalytic, Enantioselective α-Additions of Isocyanides: Lewis Base Catalyzed Passerini-Type Reactions, The Journal of Organic Chemistry, 2005, 70, 9667-9676.